MedComm | Roles of hepatic atypical protein kinase C hyperactivity and hyperinsulinemia in insulin‐resistant forms of obesity and type 2 diabetes mellitus

Open the phone and scan
Diet‐induced obesity, the metabolic syndrome, type 2 diabetes (DIO/MetS/T2DM), and their adverse sequelae have reached pandemic levels. In mice, DIO/MetS/T2DM initiation involves diet‐dependent increases in lipids that activate hepatic atypical PKC (aPKC) and thereby increase lipogenic enzymes and proinflammatory cytokines. These or other hepatic aberrations, via adverse liver‐to‐muscle cross talk, rapidly impair postreceptor insulin signaling to glucose transport in muscle. The ensuing hyperinsulinemia further activates hepatic aPKC, which first blocks the ability of Akt to suppress gluconeogenic enzyme expression, and later impairs Akt activation, further increasing hepatic glucose production. Recent findings suggest that hepatic aPKC also increases a proteolytic enzyme that degrades insulin receptors. Fortunately, all hepatic aberrations and muscle impairments are prevented/reversed by inhibition or deficiency of hepatic aPKC. But, in the absence of treatment, hyperinsulinemia induces adverse events, some by using “spare receptors” to bypass receptor defects. Thus, in brain, hyperinsulinemia increases Aβ‐plaque precursors and Alzheimer risk; in kidney, hyperinsulinemia activates the renin–angiotensin–adrenal axis, thus increasing vasoconstriction, sodium retention, and cardiovascular risk; and in liver, hyperinsulinemia increases lipogenesis, obesity, hepatosteatosis, hyperlipidemia, and cardiovascular risk. In summary, increases in hepatic aPKC are critically required for development of DIO/MetS/T2DM and its adverse sequelae, and therapeutic approaches that limit hepatic aPKC may be particularly effective.

In skeletal muscle and adipocytes, both Akt and aPKC operate downstream of IRS‐1 to mediate insulin effects on glucose transport. In cardiac muscle, whereas IRS‐1 controls aPKC and thus glucose transport during insulin action, IRS‐2 and Akt appear to be constitutively active. In liver, whereas Akt is largely controlled by IRS‐1, aPKC is controlled by IRS‐2; and, during insulin action, whereas both Akt and aPKC increase lipogenesis, Akt alone increases glucose storage in glycogen and suppresses glucose release and gluconeogenesis (Fig. 1).
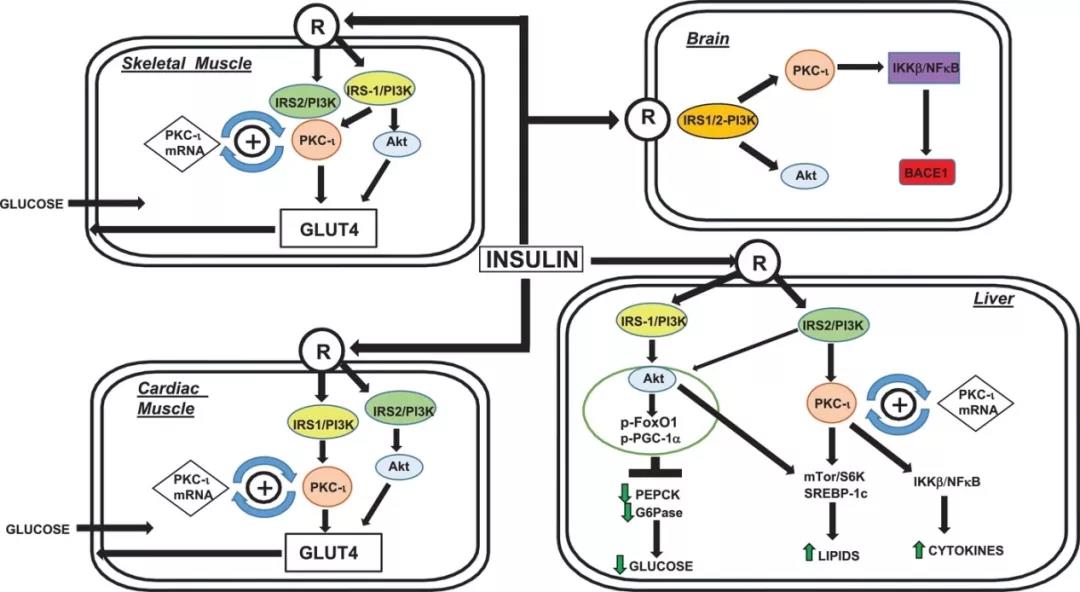
Fig. 1 Akt and aPKC mediate insulin effects on glucose transport
At this point, it seems clear that, in DIO/MetS/T2DM, hepatic aPKC hyperactivity plays a key role in producing aberrations in hepatic signaling that lead to increases in expression of gluconeogenic, lipogenic, and proinflammatory enzymes/cytokines, and subsequent development of systemic insulin resistance, glucose intolerance, and hyperinsulinemia, which in turn causes or abets development of liver‐dependent MetS features, and cardiovascular and neurological sequelae. Authors know that this sequence can be controlled by limiting hepatic aPKC. The next challenge is to develop safe therapeutic approaches that limit hepatic aPKC.
Article Access: https://onlinelibrary.wiley.com/doi/10.1002/mco2.54
Website for MedComm: https://onlinelibrary.wiley.com/journal/26882663
Looking forward to your contributions.

