Molecular Biomedicine | GMPPB-congenital disorders of glycosylation associate with decreased enzymatic activity of GMPPB

Open the phone and scan
The congenital disorders of glycosylation (CDG) are a family of metabolic diseases in which glycosylation of proteins or lipids is deficient. GDP-mannose pyrophosphorylase B (GMPPB) mutations lead to CDG, characterized by neurological and muscular defects. However, the genotype-phenotype correlation remains elusive, limiting our understanding of the underlying mechanism and development of therapeutic strategy. Here, authors report a case of an individual presenting congenital muscular dystrophy with cerebellar involvement, who presents two heterozygous GMPPB mutations (V111G and G214S). The V111G mutation significantly decreases GMPPB’s enzymatic activity. By measuring enzymatic activities of 17 reported GMPPB mutants identified in patients diagnosed with GMPPB-CDG, they discover that all tested GMPPB variants exhibit significantly decreased enzymatic activity. Using a zebrafish model, they find that Gmppb is required for neuronal and muscle development, and further demonstrate that enzymatic activity of GMPPB mutants correlates with muscular and neuronal phenotypes in zebrafish. Taken together, their findings discover the importance of GMPPB enzymatic activity for the pathogenesis of GMPPB-CDG, and shed light for the development of additional indicators and therapeutic strategy.

The patient was a 1-year old Chinese male, who was diagnosed with CMD with cerebellar involvement (CMD-CRB). His brain showed smaller size of cerebellar hemisphere and enlarged bilateral lateral ventricles, detected by MRI at his fetal stage (34 week), which indicated a possibility of cerebellar hypoplasia (Fig. 1). Postnatal brain MRI result also revealed an abnormal development of cerebellum (Fig. 1). The patient also presented with muscle weakness in upper and lower limbs as early as the age of 1 month. He gradually displayed more behavioral problems, including difficulties in raising his head and crawling. Muscle MRI showed edema of gluteus maximus (Fig. 1). Muscle biopsy indicated chronic myopathy (Fig. 1). During following-up examination, elevated creatine kinase (CK) was recorded, which fluctuated between 391 and 898 U/l (55-170 U/l) (Table 1), consistent with muscle defects.
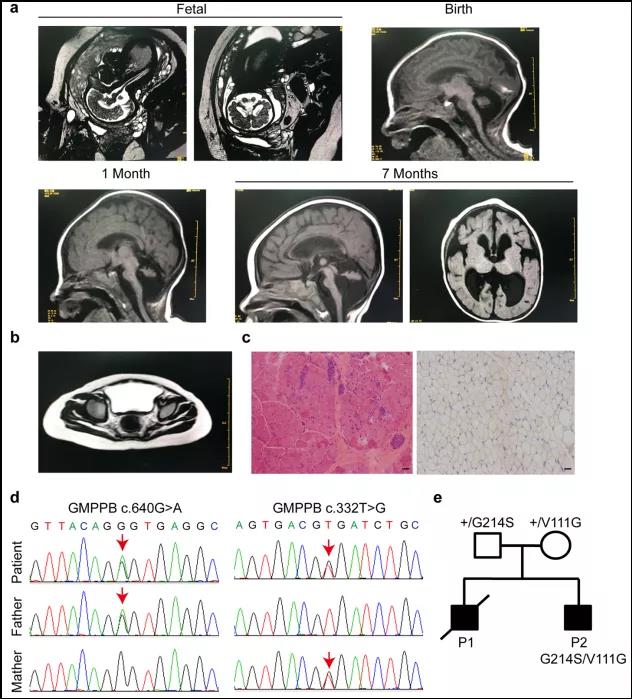
Clinical and genetic characterization
So far, more than 50 different GMPPB mutations have been reported. The clinical spectrum of GMPPB-CDG spans from CMS, LGMD to CMD with or without brain abnormalities [10, 13, 14, 18, 22]. However, the mechanism underlying this broad phenotypic spectrum remains unknown. In the present study, authors report two novel GMPPB mutations in a patient diagnosed with CMD. They identify enzymatic activity of GMPPB as a key determinant of GMPPB-CDG development. And they find that enzymatic activity of GMPPB variant correlates its ability in rescuing muscular and neuronal phenotypes in zebrafish. Their study suggests that the cellular level of GDP-mannose could correlate with patients’ symptoms.
Article Access: https://link.springer.com/article/10.1186/s43556-021-00034-3
Website for Molecular Biomedicine: https://doi.org/10.1186/s43556-021-00027-2
Looking forward to your contributions

